Using NGS for COVID-19 testing
Over the last few weeks of the COVID-19 pandemic, limited availability and throughput of the existing COVID-19 test kit has hindered our ability to monitor the global spread of the causal virus, SARS-CoV-2. As of March 24nd, the CDC reported having only tested 4,651 and 83,391 patients across the U.S. in CDC and public health laboratories, respectively, since January 18th – with the bulk of those tests done within the past week. Limited testing – frequently reserved for critical condition patients – and the increasing evidence that many infected people are asymptomatic, suggests that many cases are going undiagnosed, contributing to difficulty in containment1.
The current test kit for COVID-19 runs a Real-Time Reverse Transcriptase (RT)-PCR reaction on an Applied Biosystems 7500 Fast DX Real-Time PCR Instrument, to detect the presence of the viral RNA in up to 22 specimens at a time (see CDC Tests for COVID-19, Emergency Use Diagnostic Panel Instructions, and Lab Research Use Protocol). In collaboration with my research group, I recently devised an alternate, concept workflow that uses next-generation DNA sequencing (NGS), instead of rRT-PCR, to greatly scale the number of samples that can be assayed simultaneously. This test would work by appending unique DNA sequence “barcodes” to the nucleic acid (RNA) in each patient sample, followed by sample pooling, high-throughput next-generation sequencing, demultiplexing and counting of patient sequence reads to diagnose on the order of 10,000 patients simultaneously. This approach is naturally scalable and with a few logistics worked out could rapidly test on the order of millions of patients in the event of a major epidemic.
Here I briefly review the proposed protocol and provide my thoughts on how it can be time and cost competitive at scale. Importantly, at this time the protocol is being experimentally validated, however we are confident that it will work well as it combines the existing CDC workflow with high-throughput barcoding assays that we conduct routinely in our synthetic biology research. Our proposed workflow and supplementary data, including NGS-pipeline and designed non-repetitive DNA sequence barcodes2 will be available soon via preprint server. Of note, the Python scripts used to generate primers and sequence barcodes acts as a sort of “recipe” that can be used to design new sets of barcodes in the future, specific to any virus genome.
Testing Efforts in the U.S.
Within the past two weeks, we’ve observed a flurry of activity to address the COVID-19 testing crisis in the United States. The FDA has issued emergency use authorization (EUA) to a number of players around the country such as Roche, Thermo Fisher, and Curative (California) – to name a few – to scale and immediately begin using comparable diagnostic tests to detect SARS-CoV-2. These efforts generally use the same core assay (rRT-PCR), but with increased scale by leveraging company infrastructure, such as hiring more technicians, using automated sample handling robotics or running multiple dedicated instruments in parallel. Alternative point-of-care (POC) type assays, similar to that of pregnancy tests, are also being developed that offer to be cheap and provide a rapid readout3,4, however manufacturing and availability of these virus-specific tests may be a challenge during a rapid, wide-spread outbreak as we are currently experiencing.
Testing for virus infection involves many challenges including collecting relevant clinical specimen type(s), extracting the genetic material at-scale (see RNA Extraction Kits for COVID-19 Tests Are in Short Supply in US), obtaining sufficient genetic material with low levels of virus often present, and more. While these challenges are being investigated5 and need to be addressed immediately, a key bottlekneck to testing many patients during a major outbreak is the inherent lack of scalability of the core rRT-PCR assay.
Some approaches can be taken to partially scale the current assay. For example, combining specimen has been recommended by the CDC to be done for specimens of mixed type (e.g. nasopharyngeal and oropharyngeal swabs) to cut the need to run multiple RT-PCR assays for the same patient. Another concept, “sample pooling”, combines patient samples in small batches, that are tested together, and if the test result is negative, all patients in that batch are deemed negative for the virus, but if the result comes back positive, further testing is conducted on the individuals of that subgroup to determine which were positive6.
We were inspired by the concept of sample pooling and our experience using barcoding sequence variants quantified with NGS7-9, to come up with the following workflow.
Concept Workflow
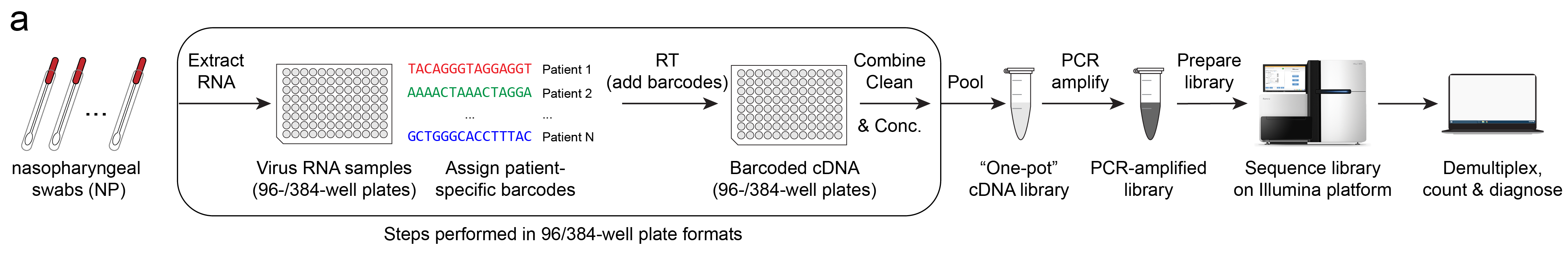
The workflow is as follows (illustrated above):
- RNA is extracted from patient specimens*.
- Unique barcodes as assigned to each patient.
- Barcoded cDNA is created using Reverse-Transcription (RT).
- Samples are cleaned, concentrated and pooled together**.
- cDNA is amplified with PCR.
- Amplified library is size selected and prepared for sequencing.
- Library is sequenced via next-generation sequencing (NGS)***.
- Reads are demultiplexed for each barcode (patient) and counted.
A diagnosis is then determined by the number of read counts of virus RNA and control RNAs. Without going into too much detail, simply put, for a given barcode, if reads matching the expected virus sequence are present, the test result is positive.
*Step 1, RNA extractions, may be optional and alternatives are currently being investigated by other research groups.
**Steps 1-4 are done in multi-well plate format, optionally with liquid handling robots to greatly increase the processing speeds of these steps.
***Sequencing platform may be Illumina or Oxford Nanopore (MinION). I will go into details between these in a future post.
Understanding Barcodes
Barcodes are unique DNA sequences designed to identify a variant or a sample in a pool of many. Much like the barcodes you can find on produce in the grocery market, genetic barcodes can be read by a machine, a sequencer. While traditional barcodes are composed of black parallel lines of variable widths, DNA barcodes are instead defined by their nucleotide sequence. Natural DNA consists of four nucleotides: A, T, C, and G. This means at each position in a barcode, you have four unique choices, and for barcodes of length N, you have 4N unique possible sequences to choose from.
For example, here are three 15-nucleotide DNA barcodes:
ID | sequence
BC1 | AGCTATCGTAGCTTG
BC2 | GGAGGAAACCCACCC
BC3 | GTAAAAATTTACAGA
The longer the barcodes, the more possible sequences you can choose from and the more possible differences there can be between any two. It turns out designing non-repetitive genetic parts, such as barcodes, within a constrained sequence space is a fascinating problem in itself, one our group has explored2.
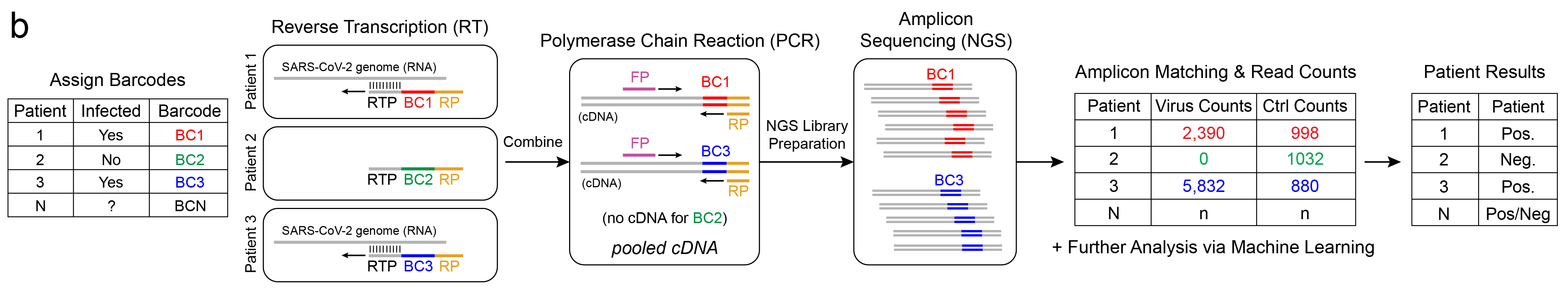
Barcodes allow us to “tag” independent samples, pool them together (multiplex), sequence, then tease apart individual results (demultiplex). For example, let’s say we have 3 patients: Patients 1, 2, and 3, and we assign them barcodes: BC1, BC2, and BC3, respectively. Let’s say Patients 1 & 3 are infected with SARS-CoV-2 and Patient 2 is not. The workflow uses unique barcoded RT primers for the Reverse Transcription reaction to generate cDNA. Note that since Patient 2 is negative for SARS-CoV-2, and no virus RNA is present in that sample, the RT primer has nothing to bind to and no cDNA-BC2 is synthesized.
The barcoded cDNA is then pooled together and a common pair of forward and reverse primers are used to amplify the one-pot DNA using Polymerase Chain Reaction (PCR). You may notice this workflow just used RT and PCR. This is an important point. Because this workflow also uses RT-PCR, just not real-time, we can expect similar sensitivity to rRT-PCR, that is, the limit-of-detection is expected to be similar. There is a notion too that RNA-seq (NGS) is not as accurate as rRT-PCR (qPCR), but from our experience, as long as the preprocessing steps are the same and there is sufficient sequencing depth, they yield equivalent results (R2=0.98)10.
Last, the library is sequenced, and a computational pipeline counts occurance of each barcode. The barcoded sequences, or amplicons, are confirmed to match the expected sequence and a count is incremented for the barcode, or corresponding patient. Depending on the counts for virus sequence and other controls, the patients are assigned a test result: positive, negative, inconclusive, or invalid.
Additional Multiplexing
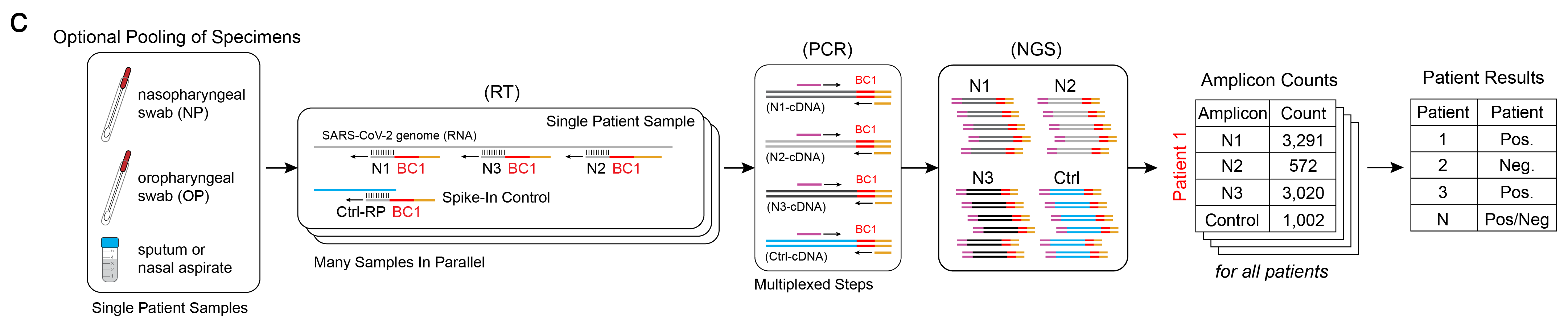
One strength of this workflow is that additional components are naturally multiplexable. If multiple patient samples are taken – say, spit and swab samples – they can be combined and the RNA may be extracted together to increase the amount of starting genetic material. Additionally, multiple RT primers may be used together for various target sites on the virus genome and for controls, such as a human RNA control or spike-in DNA controls. In other words, these individual amplicons, which are assayed individually in the rRT-PCR workflow, are assayed together here in ours (above). The result of the workflow are tabulated counts for each unique targeted amplicon, per patient. Beyond these benefits, Illumina sequencers designed for higher throughput can also run multiple libraries simultaneously, meaning sequencing could be done for tens of thousands of patients simultaneously.
Time and Cost Considerations
Cost – Running the proposed assay would require around $5-10k worth of library prep and sequencing costs on an Illumina platform, and if the assay is run for 10,000 patients, this portion of the workflow would cost a fraction of a dollar per patient – this is the economy of scale gained by multiplexing. In comparison, the current workflow requires relatively expensive rRT-PCR reagents and probes which are consumed 3 units (reactions) worth per patient.
Time – The proposed workflow could be executed anywhere between 2-5 days, depending on the degree of automation. A 3 day window seems realistic, provided there are dedicated Illumina sequencers. In comparison, the current rRT-PCR workflow takes about a day to complete. We consider a two day lag acceptable since many viruses have long incubation periods and patients (symptomatic or not) would likely already be self isolating in the window that the results are returned.
Disclaimers
As we prototype this concept, I hope to share posts describing our efforts and progress. Realistically, this diagnostic approach could be fully validated and possibly approved by the fall.
If you want to find out more about COVID-19 and get the latest information on the pandemic, please visit the WHO website at https://www.who.int/.
If you’re in the United States and have questions about development and performance of diagnostic tests for SARS-CoV-2, please visit FAQs on Diagnostic Testing for SARS-CoV-2.
References
-
Li, R., Pei, S., Chen, B., Song, Y., Zhang, T., Yang, W., & Shaman, J. (2020). Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV2). Science.
-
Hossain, A., Lopez, E., Halper, S. M., Cetnar, D. P., Reis, A. C., Strickland, D., Klavins, E., Salis, H. M. (2020). Automated design of thousands of highly non-repetitive genetic parts for engineering evolutionary robust genetic systems. Accepted.
-
Myhrvold, C., Freije, C. A., Gootenberg, J. S., Abudayyeh, O. O., Metsky, H. C., Durbin, A. F., … & Garcia, K. F. (2018). Field-deployable viral diagnostics using CRISPR-Cas13. Science, 360(6387), 444-448.
-
Broughton, J. P., Deng, X., Yu, G., Fasching, C. L., Singh, J., Streithorst, J., … & Hsu, E. (2020). Rapid Detection of 2019 Novel Coronavirus SARS-CoV-2 Using a CRISPR-based DETECTR Lateral Flow Assay. medRxiv.
-
Bruce, A. E., Tighe, S., et al. (2020). RT-qPCR detection of SARS-CoV-2 RNA from patient nasopharyngeal swab using QIAGEN RNEasy kits or directly via omission of an RNA extraction step. biorxiv.
-
Bilder, C. R., & Tebbs, J. M. (2012). Pooled‐testing procedures for screening high volume clinical specimens in heterogeneous populations. Statistics in medicine, 31(27), 3261-3268.
-
Knapp, M., Stiller, M., & Meyer, M. (2012). Generating barcoded libraries for multiplex high-throughput sequencing. In Ancient DNA (pp. 155-170). Humana Press.
-
Kosuri, S., Goodman, D. B., Cambray, G., Mutalik, V. K., Gao, Y., Arkin, A. P., … & Church, G. M. (2013). Composability of regulatory sequences controlling transcription and translation in Escherichia coli. Proceedings of the National Academy of Sciences, 110(34), 14024-14029.
-
Goodman, D. B., Church, G. M., & Kosuri, S. (2013). Causes and effects of N-terminal codon bias in bacterial genes. Science, 342(6157), 475-479.
-
Reis, A. C., Halper, S. M., Vezeau, G. E., Cetnar, D. P., Hossain, A., Clauer, P. R., & Salis, H. M. (2019). Simultaneous repression of multiple bacterial genes using nonrepetitive extra-long sgRNA arrays. Nature biotechnology, 37(11), 1294-1301.